Epigenetic aging. In 2013 we showed that large parts of the methylome are remodeled with age, a process that is accelerated by disease and slowed in certain genotypes and in women versus men. These findings led to the first “epigenetic clock” model for predicting rate of biological aging (Hannum et al. Cell. 2013). We have since reported that these changes are accelerated by viral infection (Gross et al. Mol. Cell. 2016) and slowed by anti-aging treatments such as caloric restriction and rapamycin (Wang et al. Genome Biology 2017). Most recently, we used epigenetic profiles to translate age between humans and dogs (Wang et al., Cell Systems 2020). Comparison of Labrador retriever and human methylomes revealed a nonlinear relationship between dog and human aging which did not follow the conventional wisdom that 1 dog year = 7 human years, leading to a story that was popularized by many news outlets.

Tina Wang, et al. Quantitative Translation of Dog-to-Human Aging by Conserved Remodeling of the DNA Methylome. Cell Systems (2020). [PDF] [PubMed]
Selected Press:
- Old Dogs, New Research and the Secrets of Aging. James Gorman, New York Times (9 Nov 2020) [PDF]
- Podcast: How old is your dog in human years? Genetic study offers a new way to answer that question. Scientific American (9 January 2020)
- How to calculate your dog’s real age. BBC (6 Jan 2020)
- Radio Interview: Measuring A Dog’s Age Is More Complicated Than You Think. KCBS Radio. (14 Dec 2019)
- A New Way To Calculate Your Dog’s Age. NPR (7 Dec 2019)
- Scientists have come up with a better way to convert your dog’s age to human years. Washington Post (28 Nov 2019)
- Convert your dog’s age into human years using this new formula. New Scientist (21 Nov 2019)
- Science May Have a Better Way to Translate Dog Years to Human Years. Discover Magazine (20 Nov 2019)
- Calculate Your Dog’s Age With This New, Improved Formula. Smithsonian Magazine (19 Nov 2019)
- There’s a New Way to Calculate Your Dog’s Age in Human Years. New York Magazine, The Cut (19 Nov 2019)
- There’s a Better Way Calculate Your Dog’s True Age in Dog Years, Researchers Say. People Magazine (19 Nov 2019)
- The New Formula to Calculate Your Dog’s Age in Human Years. Popular Mechanics (18 Nov 2019)
- How old is your dog REALLY in ‘human years’? Researchers come up with a new formula for calculating the ‘true age’ of your canine friend (and it’s not multiplying it by seven). Daily Mail UK (18 Nov 2019)
- Here’s a better way to convert dog years to human years, scientists say. Science Magazine (15 Nov 2019)
In 2012, in collaboration with the laboratory of Drs. Kang Zhang (UCSD) and Stephen Friend (Sage), we built a quantitative model of aging using measurements at more than 450,000 methylation (CpG) markers from the whole blood of 656 human individuals, aged 19 to 101. This model was able to very accurately predict age from the state of the methylome, which we showed was impacted by gender and genetic variants. We found our aging model was upheld in other human tissues, and that it revealed an advanced aging rate in tumor tissue [Hannum et al. Molecular Cell 2013].
More recently, we sought to advance the field of epigenetic aging in two ways. First, we partnered with Dr. Howard Fox (formerly TSRI, now University of Nebraska) to explore the effects of viral infection on epigenetic aging rate. In our study, HIV+ men on anti-retroviral therapy had significantly advanced epigenetic aging versus controls [Gross et al. Molecular Cell 2016]. Second, we worked jointly with Dr. Peter Adams (Sanford Burnham Prebys) to study the effects of pro-longevity interventions on epigenetic age in mice. Rapamycin treatment decreased the rate of epigenetic aging, as did caloric restriction and the genetic condition of dwarfism [Wang et al. Genome Biology 2017].
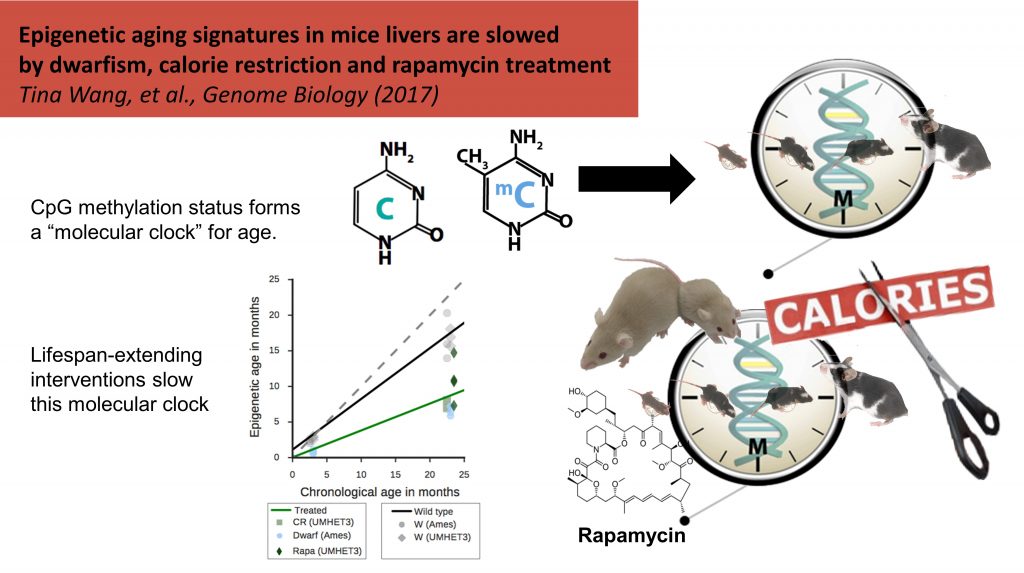
[Wang et al. Genome Biology 2017]
We then used epigenetic profiles to translate age between humans and dogs [Wang et al., Cell Systems (2020) and BioRxiv (2019)]. Mammals progress through similar physiological stages during life, from early development to puberty, aging, and death. Yet, the extent to which this conserved physiology reflects conserved molecular events is unclear. Here, we map common epigenetic changes experienced by mammalian genomes as they age, focusing on evolutionary comparisons of humans to dogs, an emerging model of aging. Using targeted sequencing, we characterize the methylomes of 104 Labrador retrievers spanning a 16 year age range, achieving >150X coverage within mammalian syntenic blocks. Comparison with human methylomes reveals a nonlinear relationship which translates dog to human years, aligns the timing of major physiological milestones between the two species, and extends to mice. Conserved changes center on specific developmental gene networks which are sufficient to capture the effects of anti-aging interventions in multiple mammals. These results establish methylation not only as a diagnostic age readout but as a cross-species translator of physiological aging milestones.
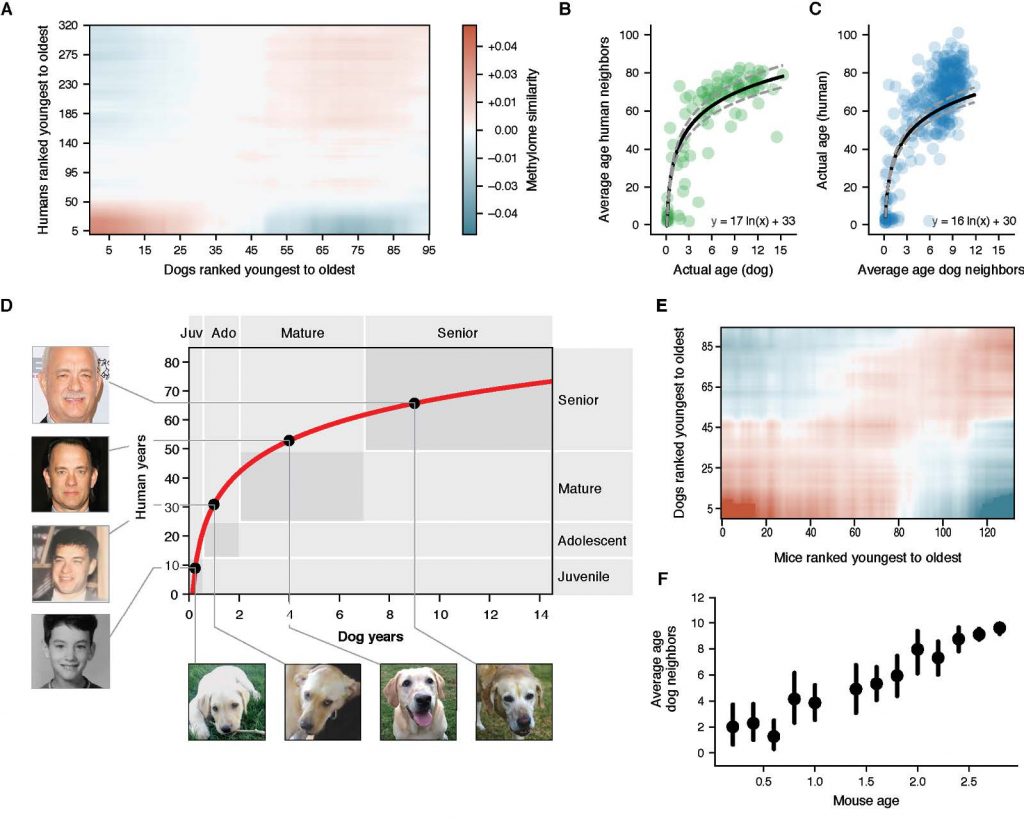
Figure 2: A non-linear transformation from dog to human age. (Wang et al. Cell Systems (2020))