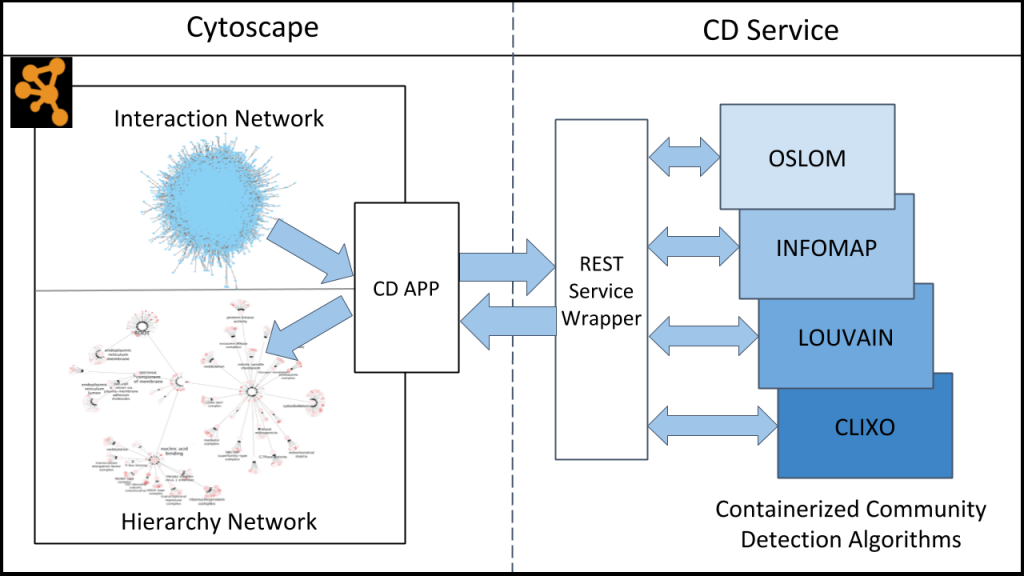
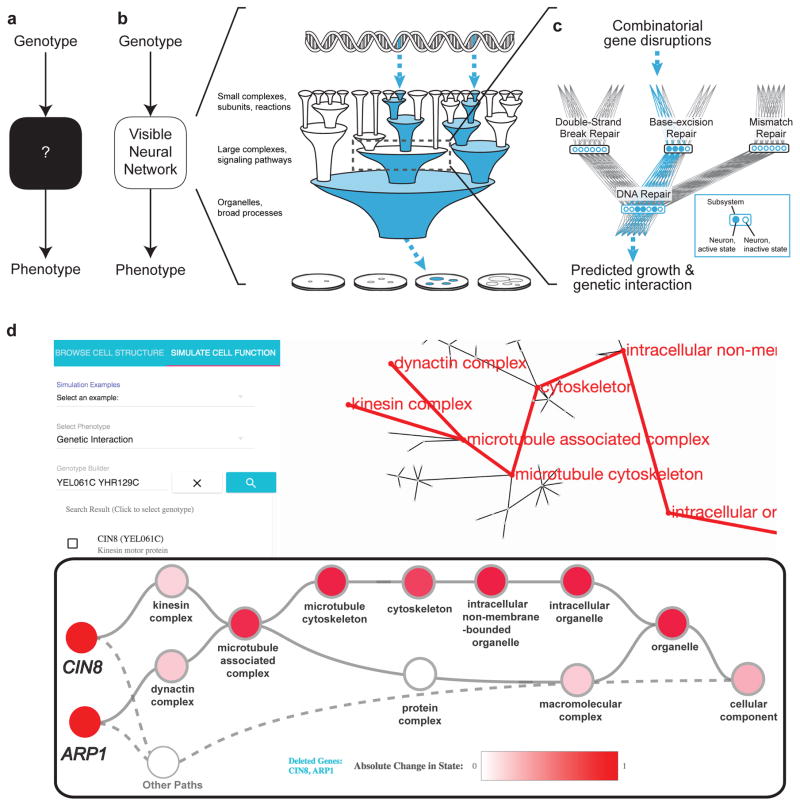
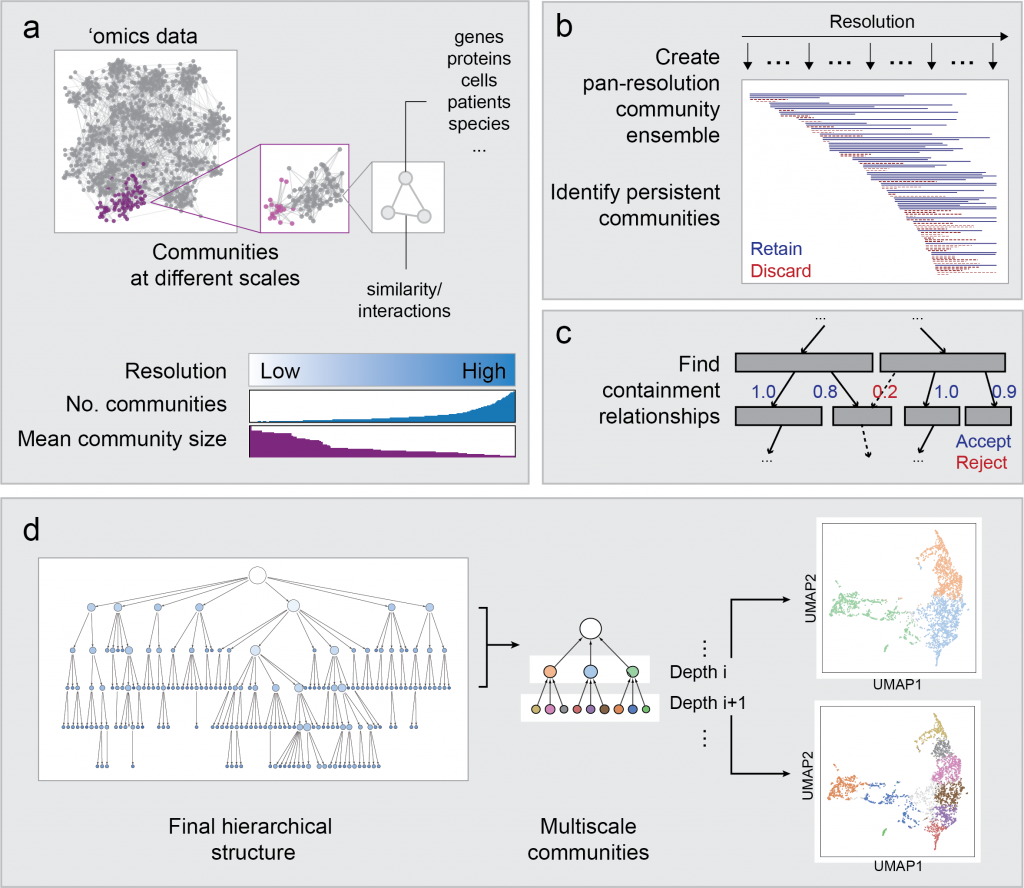
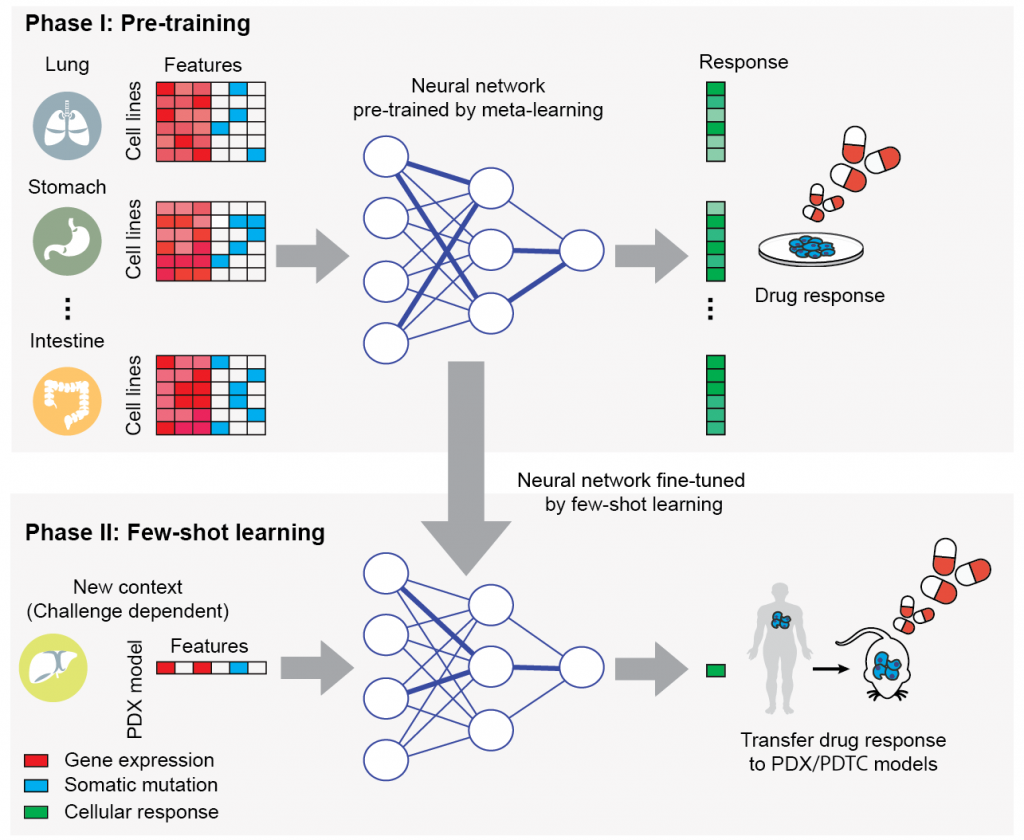
Legacy Software
Cell Circuit Search: Molecular interaction models provide us with a framework for integrating the large-scale data that we are now able to collect at multiple levels of biological information – genes, RNAs, proteins, and small molecules. Cell Circuit Search is a web-based interface for searching for genes that appear in our library of network models.
NetworkBLAST Software: NetworkBlast analyzes protein interaction networks in order to predict previously unknown relationships. It can compare multiple species’ protein interaction networks and infer interactions through homology. The program is best used in conjunction with Cytoscape to easily visualize the returned data.
PathBLAST Website: Pathway alignment and query against protein interaction databases to identify conserved protein interaction networks between species. PathBLAST searches the protein-protein interaction network of the target organism to extract all protein interaction pathways that align with a pathway query.
VERA and SAM: VERA and SAM was developed to address the need for a better statistical test for identifying differentially-expressed genes. VERA estimates the parameters of a statistical model that describes multiplicative and additive errors influencing an array experiment, using the method of maximum likelihood. SAM gives a value, lambda, for each gene on an array, which describes how likely it is that the gene is expressed differently between the two cell populations and was developed to address the need for a better statistical test for identifying differentially-expressed genes.
Dapple: Dapple is a program for quantitating spots on a two-color DNA microarray image. Given a pair of images from a comparative hybridization, Dapple finds the individual spots on the image, evaluates their qualities, and quantifies their total fluorescent intensities. Dapple is designed to work with microarrays on glass and is a program for quantitating spots on a two-color DNA microarray image.
enoLOGOS: Program enoLOGOS generates LOGOs of transcription factor DNA binding sites from various types of input matrices. It can utilize standard count matrices, probability matrices or matrices of “energy” values (i.e., log-frequencies).