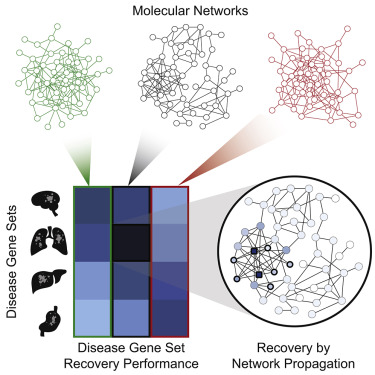
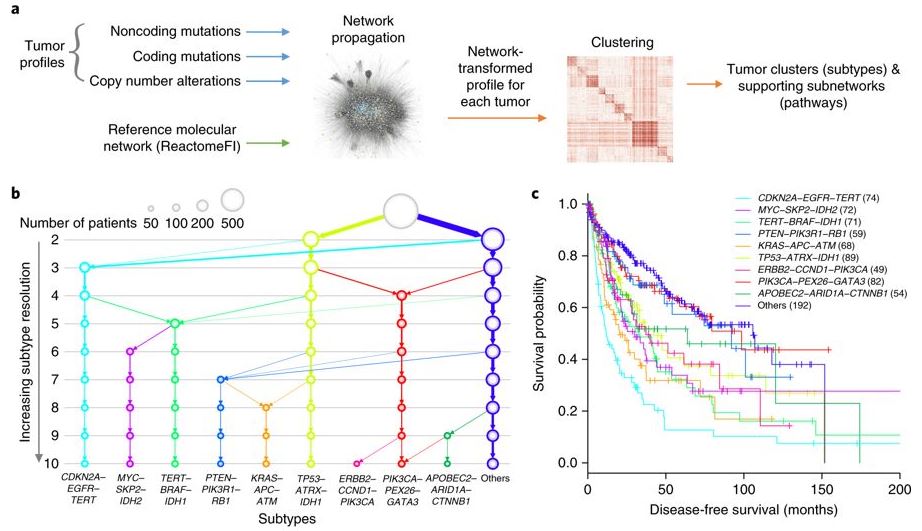
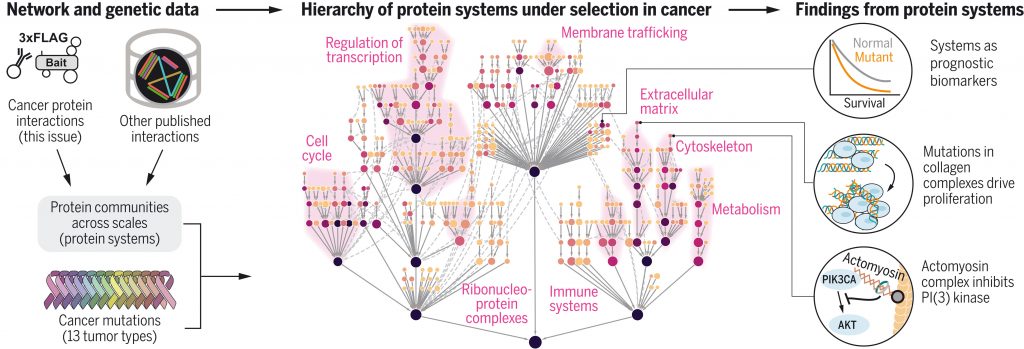
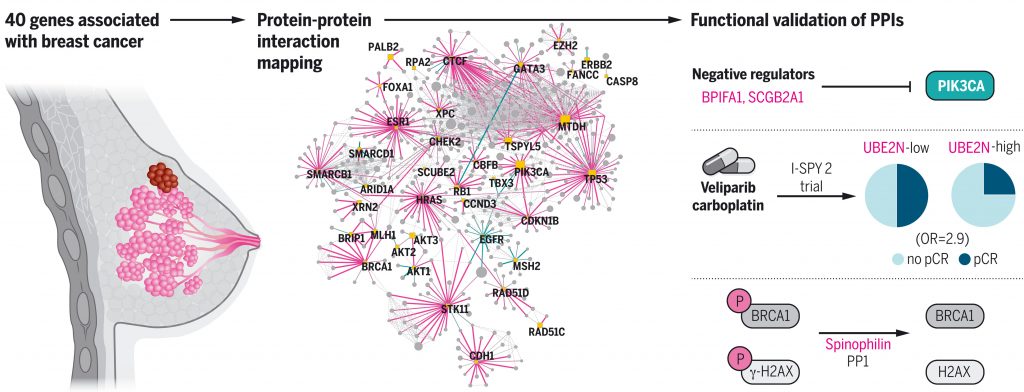
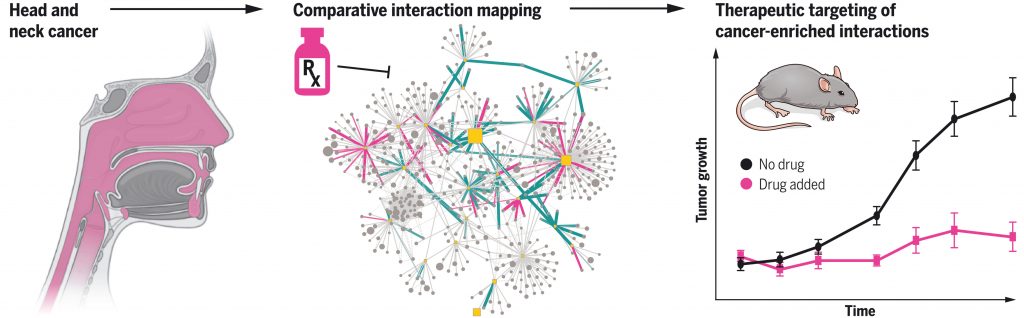
Highlights from the past few years include a study of epistatic interactions among the mutations found in tumor genomes (Van de Haar et al. Cell 2019) as well as a review, with Jonathan Flint, providing guidelines for use of molecular networks in studies of genome-wide association for psychiatric disorders (Flint and Ideker, PLoS Genetics, 2019). Finally, I am co-corresponding author on three papers (together with Nevan Krogan) which comprehensively map the protein networks underlying multiple tumor types and use these maps to identify >300 protein complexes and larger protein assemblies under mutational selection in cancer. This new work includes published comprehensive protein interaction networks for breast cancer (Kim et al. Science 2021) and head-and-neck cancer (Swaney et al., Science 2021). A third paper analyzes these and other network data to identify a large compendium of protein complexes under selective pressure for mutation (Zheng et al. Science 2021). These papers were published as back-to-back articles in the same issue; they were the result of a more than five-year collaboration between my laboratory and that of Nevan Krogan at UCSF, with significant contributions from the laboratories of Silvio Gutkind (Chair of UCSD Pharmacology), Stephanie Fraley (UCSD Bioengineering) and others.